
| 產(chǎn)品編號 | bsm-52505R |
| 英文名稱 | Rabbit Anti-PRNP antibody |
| 中文名稱 | 朊病毒蛋白CD230重組兔單抗 |
| 別 名 | Alternative prion protein; AltPrP; ASCR; CD230; CD230 antigen; CJD; GSS; KURU; Major prion protein; p27 30; PRIO_HUMAN; Prion related protein; PRIP; PrP; PrP27 30; PrP27-30; PrP33-35C; PrPC; PrPSc; Sinc |
| 研究領(lǐng)域 | 細(xì)胞生物 神經(jīng)生物學(xué) 干細(xì)胞 細(xì)菌及病毒 細(xì)胞表面分子 |
| 抗體來源 | Rabbit |
| 克隆類型 | Recombinant |
| 交叉反應(yīng) | Human,Mouse,Rat |
| 產(chǎn)品應(yīng)用 | WB=1:500-5000,IHC-P=1:100-500,IHC-F=1:400-800,IF=1:50-200
not yet tested in other applications. optimal dilutions/concentrations should be determined by the end user. |
| 理論分子量 | 25kDa |
| 細(xì)胞定位 | 細(xì)胞核 細(xì)胞漿 細(xì)胞膜 |
| 性 狀 | Liquid |
| 濃 度 | 1mg/ml |
| 免 疫 原 | KLH conjugated synthetic peptide derived from PRNP: 161-253 / 253 |
| 亞 型 | IgG |
| 純化方法 | affinity purified by Protein A |
| 緩 沖 液 | 0.01M TBS (pH7.4) with 1% BSA, 0.02% Proclin300 and 50% Glycerol. |
| 保存條件 | Shipped at 4℃. Store at -20℃ for one year. Avoid repeated freeze/thaw cycles. |
| 注意事項(xiàng) | This product as supplied is intended for research use only, not for use in human, therapeutic or diagnostic applications. |
| PubMed | PubMed |
| 產(chǎn)品介紹 |
The function of PrP is still under debate. May play a role in neuronal development and synaptic plasticity. May be required for neuronal myelin sheath maintenance. May play a role in iron uptake and iron homeostasis (By similarity). Isoform 2 may act as a growth suppressor by arresting the cell cycle at the G0/G1 phase. Soluble oligomers are toxic to cultured neuroblastoma cells and induce apoptosis (in vitro). Function: The function of PrP is still under debate. May play a role in neuronal development and synaptic plasticity. May be required for neuronal myelin sheath maintenance. May play a role in iron uptake and iron homeostasis (By similarity). Isoform 2 may act as a growth suppressor by arresting the cell cycle at the G0/G1 phase. Soluble oligomers are toxic to cultured neuroblastoma cells and induce apoptosis (in vitro). Subunit: Monomer and homodimer. Has a tendency to aggregate into amyloid fibrils containing a cross-beta spine, formed by a steric zipper of superposed beta-strands. Soluble oligomers may represent an intermediate stage on the path to fibril formation. Copper binding may promote oligomerization. Interacts with GRB2, APP, ERI3/PRNPIP and SYN1. Mislocalized cytosolically exposed PrP interacts with MGRN1; this interaction alters MGRN1 subcellular location and causes lysosomal enlargement (By similarity). Interacts with KIAA1191. Subcellular Location: Cell membrane. Golgi apparatus and Cytoplasm. Nucleus. Accumulates outside the secretory route in the cytoplasm, from where it relocates to the nucleus. Post-translational modifications: The glycosylation pattern (the amount of mono-, di- and non-glycosylated forms or glycoforms) seems to differ in normal and CJD prion. Isoform 2 is sumoylated by SUMO1. DISEASE: Note=PrP is found in high quantity in the brain of humans and animals infected with neurodegenerative diseases known as transmissible spongiform encephalopathies or prion diseases, like: Creutzfeldt-Jakob disease (CJD), fatal familial insomnia (FFI), Gerstmann-Straussler disease (GSD), Huntington disease-like type 1 (HDL1) and kuru in humans; scrapie in sheep and goat; bovine spongiform encephalopathy (BSE) in cattle; transmissible mink encephalopathy (TME); chronic wasting disease (CWD) of mule deer and elk; feline spongiform encephalopathy (FSE) in cats and exotic ungulate encephalopathy (EUE) in nyala and greater kudu. The prion diseases illustrate three manifestations of CNS degeneration: (1) infectious (2) sporadic and (3) dominantly inherited forms. TME, CWD, BSE, FSE, EUE are all thought to occur after consumption of prion-infected foodstuffs. Defects in PRNP are the cause of Creutzfeldt-Jakob disease (CJD) [MIM:123400]. CJD occurs primarily as a sporadic disorder (1 per million), while 10-15% are familial. Accidental transmission of CJD to humans appears to be iatrogenic (contaminated human growth hormone (HGH), corneal transplantation, electroencephalographic electrode implantation, etc.). Epidemiologic studies have failed to implicate the ingestion of infected annimal meat in the pathogenesis of CJD in human. The triad of microscopic features that characterize the prion diseases consists of (1) spongiform degeneration of neurons, (2) severe astrocytic gliosis that often appears to be out of proportion to the degree of nerve cell loss, and (3) amyloid plaque formation. CJD is characterized by progressive dementia and myoclonic seizures, affecting adults in mid-life. Some patients present sleep disorders, abnormalities of high cortical function, cerebellar and corticospinal disturbances. The disease ends in death after a 3-12 months illness. Defects in PRNP are the cause of fatal familial insomnia (FFI) [MIM:600072]. FFI is an autosomal dominant disorder and is characterized by neuronal degeneration limited to selected thalamic nuclei and progressive insomnia. Defects in PRNP are the cause of Gerstmann-Straussler disease (GSD) [MIM:137440]. GSD is a heterogeneous disorder and was defined as a spinocerebellar ataxia with dementia and plaquelike deposits. GSD incidence is less than 2 per 100 million live births. Defects in PRNP are the cause of Huntington disease-like type 1 (HDL1) [MIM:603218]. HDL1 is an autosomal dominant, early onset neurodegenerative disorder with prominent psychiatric features. Defects in PRNP are the cause of kuru (KURU) [MIM:245300]. Kuru is transmitted during ritualistic cannibalism, among natives of the New Guinea highlands. Patients exhibit various movement disorders like cerebellar abnormalities, rigidity of the limbs, and clonus. Emotional lability is present, and dementia is conspicuously absent. Death usually occurs from 3 to 12 month after onset. Defects in PRNP are the cause of spongiform encephalopathy with neuropsychiatric features (SENF) [MIM:606688]; an autosomal dominant presenile dementia with a rapidly progressive and protracted clinical course. The dementia was characterized clinically by frontotemporal features, including early personality changes. Some patients had memory loss, several showed aggressiveness, hyperorality and verbal stereotypy, others had parkinsonian symptoms. Prion diseases, or transmissible spongiform encephalopathies (TSEs), are manifested as genetic, infectious or sporadic, lethal neurodegenerative disorders involving alterations of the prion protein (PrP). Characteristic of prion diseases, cellular PrP (PrPc) is converted to the disease form, PrPSc, through alterations in the protein folding conformations. PrPc is constitutively expressed in normal adult brain and is sensitive to proteinase K digestion, while the altered PrPSc conformation is resistant to proteases, resulting in a distinct molecular mass after PK treatment. Consistent with the transient infection process of prion diseases, incubation of PrPc with PrPSc both in vitro and in vivo produces PrPc that is resistant to protease degradation. Infectious PrPSc is found at high levels in the brains of animals affected by TSEs, including scrapie in sheep, BSE in cattle and Cruetzfeldt-Jakob disease in humans. Similarity: Belongs to the prion family. SWISS: P04156 Gene ID: 5621 |
| 產(chǎn)品圖片 |

Blocking buffer: 5% NFDM/TBST
Primary Ab dilution: 1:1000
Primary Ab incubation condition: 4℃
overnight
Secondary Ab: Goat Anti-Rabbit IgG H&L
(HRP)
Lysate: 1: Mouse brain, 2: Rat brain
Protein loading quantity: 20 μg
Exposure time: 60 s
Predicted MW: 28 kDa
Observed MW: 25-30 kDa

Tissue: Rat cerebrum
Section type: Formalin-fixed & Paraffin -
embedded section
Retrieval method: High temperature and high
pressure
Retrieval buffer: Tris/EDTA buffer, pH 9.0
Primary Ab dilution: 1:1000
Primary Ab incubation condition: 1 hour at
room temperature
Secondary Ab: Anti-Rabbit and Mouse
Polymer HRP (Ready to use)
Counter stain: Hematoxylin (Blue)
Comment: Color brown is the positive signal
for
bsm-52505R

Tissue: Human cerebrum
Section type: Formalin-fixed & Paraffin -
embedded section
Retrieval method: High temperature and high
pressure
Retrieval buffer: Tris/EDTA buffer, pH 9.0
Primary Ab dilution: 1:1000
Primary Ab incubation condition: 1 hour at
room temperature
Secondary Ab: Anti-Rabbit and Mouse
Polymer HRP (Ready to use)
Counter stain:
Hematoxylin (Blue)
Comment: Color brown is the positive signal
for Tissue: Human cerebrum
Section type: Formalin-fixed & Paraffin -
embedded section
Retrieval method: High temperature and high
pressure
Retrieval buffer: Tris/EDTA buffer, pH 9.0
Primary Ab dilution: 1:1000
Primary Ab incubation condition: 1 hour at
room temperature
Secondary Ab: Anti-Rabbit and Mouse
Polymer HRP (Ready to use)
Counter stain:
Hematoxylin (Blue)
Comment: Color brown is the positive signal
for
bsm-52505R
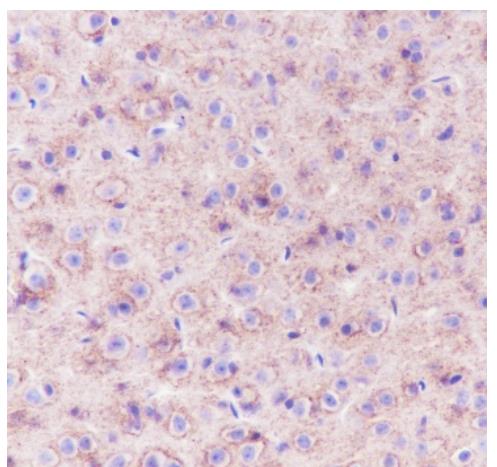
Tissue: Mouse cerebrum
Section type: Formalin-fixed & Paraffin -
embedded section
Retrieval method: High temperature and high
pressure
Retrieval buffer: Tris/EDTA buffer, pH 9.0
Primary Ab dilution: 1:1000
Primary Ab incubation condition: 1 hour at
room temperature
Secondary Ab: Anti-Rabbit and Mouse
Polymer HRP (Ready to use)
Counter stain: Hematoxylin (Blue)
Comment: Color brown is the positive signal
for bsm-52505R
|




